Enzim kinetiği

Enzim kinetiği enzimler tarafından katalizlenen kimyasal reaksiyonların bilmidir. Enzim kinetiğinde reaksiyon hızı ölçülür ve reaksiyon şartlarını değiştirmenin etkisi araştırılır. Bir enzimin kinetiğinin bu şekilde çalışılması enzimin katalitik mekanizmasını, metabolizmadaki rolünü, aktivitesinin nasıl kontrol edildiğini ve bir ilaç veya zehrin enzimi nasıl inhibe edebileceğini ortaya koyabilir.
Enzimler genelde diğer molekülleri (enzimin substratları) manipüle eden protein molekülleridir. Bu hedef moleküller enzimin aktif bölgesine bağlanır ve, enzim mekanizması denen bir seri adımlar sonucunda, ürünlere dönüşür. Bu mekanizmalar tek substratlı ve çok substratlı mekanizmalar olarak ayrılabilir. Tek bir substrata bağlanan enzimler (triozfosfat izomeraz gibi) üzerinde yapılan kinetik çalışmalar, enzimin substratına bağlanma afinitesini (ilgisini) ve devir hızını ölçmeyi amaçlar.
Enzimler birden çok substrata bağlanınca, (sağda gösterilen) dihidrofolat redüktaz gibi, enzim kinetiği bu substratların bağlanma sırasını ve ürünlerin salınma sırasını gösterebilir. Bir tek substrata bağlanıp birden çok ürün salan enzimlere örnek, proteazlardır, bunlar bir protein substratı kesip iki polipeptit ürün oluştururlar. Başka enzimler iki substratı birleştirir, öreneğin DNA polimeraz'ın DNA'ya bir nükleotit eklemesi gibi. Bu mekanizmalar genelde karmaşık bir adımlar dizisi olsa da, tipik olarak bir hız belirleyici basamak, tüm reaksiyonun hızını belirler. Bu hız belirleyici basamak kimyasal bir reaksiyon veya, enzim veya substratlarında konformasyonel bir değişim olabilir, ürünlerin enzimden salınması sırasında görüldüğü gibi.
Enzimin yapısının bilinmesi kinetik verilerin yorumlanmasında faydalıdır. Örneğin, yapının bilinmesi sayesinde substrat ve ürünlerin kataliz sırasında nasıl bağlandıkları, reaksiyon sırasından hangi değişikliklerin meydana geldiği, ve hatta mekanizmadaki belli amino asit kalıntılarınını rolleri tahmin edilebilir. Bazı enzimler mekanizma sırasında önemli derecede biçimlerini değiştirirler; böylesi durumlarda enzimin tek başına yapısının ve enzimatik reaksiyona girmeyen substrat analogları ona bağlı ikenki yapısının belirlenmesi yararlı olur.
Tüm biyolojik katalizörler protein değildir; ribozim ve ribozom gibi RNA-temelli katalizörler pek çok hücresel işlev için esastırlar, örneğin RNA uçbirleştirmesi (splicing) ve çeviri (translasyon) gibi. Ribozimler ve enzimler arasındaki temel fark, RNA katalizörlerin nükleotitlerden, enzimlerin ise amino asitlerden oluşmasıdır. Ribozimler daha sınırlı bir reaksiyonlar grubunu katalizler, ama reaksiyon mekanizmaları ve kinetikleri protein enzimleri ile aynı yöntemlerle analiz edilebilir ve sınıflandırılabilir.
Genel ilkeler
.svg.png)
Bir enzim tarafından katalizlenen reaksiyon, katalizlenmeyen reaksiyon ile aynı denge özelliklerine sahiptir. Diğer katalizörler gibi, enzimler substrat ve ürünler arasındaki dengenin konumunu değiştirmezler.[1] Ancak, katalizlenmeyen bir kimyasal reaksiyondan farklı olarak, enzimle katalizlenen reaksiyonlar doyum kinetiği gösterirler. Belli bir enzim konsantrasyonu ve nispeten düşük substrat konsantrasyonu için, reaksiyon hızı substrat konsantrasyonu ile doğrusal orantılı olarak artar; enzim moleküllerinin çoğu, reaksiyonu katalizlemek için müsaittir ve substrat konsantrasyonun artırılması, enzim ve substrat moleküllerinin birbirine rastlama hızının artması demektir. Ancak, nispeten yüksek substrat konsantrasyonlarıda, reasiyon hızı asimptotik şekilde teorik maksimuma yaklaşır; enzim aktif bölgelerinin hemen hepsi doludur ve reaksiyon hızını, enzimin kendine has (içsel) devir hızı belirler. Bu iki sınır durumun ortasındaki substrat konsantrasyonu KM olarak belirtilir. Bir diğer deyişle, en yüksek reaksiyon hızının (Vmax) yarısının elde edildiği substrat konsantrasyonu KM olarak tanımlanır.
Bir enzimin en önemli iki kinetik özelliği, enzimin belli bir substrat ile ne kadar çabuk doyuma ulaştığı ve ulaşabildiği en hızlı reaksiyon hızıdır. Bu özelliklerin bilinmesi bir enzimin hücre içinde ne yaptığını ve o şartlardaki değişiklere nasıl tepki verdiği hakkında fikir verir.
Enzim ölçümleri
Enzim ölçümleri enzim reakasiyonunun hızını ölçen laboratuvar yöntemleridir. Enzimler katalizledikleri reaksiyonlar tarafından tüketilmediği için, enzim ölçümlerinde reaksiyon hızını takip etmek için genelde ya substrat konsantrasyonu ya da ürün konsantrasyonu izlenir. Ölçüm için pek çok yöntem vardır. Spektrofotometrik ölçümlerde substrat ve ürünlerdeki ışık absorbansındaki değişim gözlemlenir; radyometrik ölçümlerde radyoaktivitenin bir ürüne dahil oluşu veya substrattan ayrılmasına bakarak zamana göre ürün oluşumunu ölçülür. Spektrofotometrik ölçümler en kullanışlıdır, çünkü reaksiyon hızının sürekli olarak ölçülmesi mümkündür. Radyometrik ölçümler numunelerin alınıp onların ölçümünü gerektirse de (yani kesintili ölçümlerdir) genelde daha duyarlıdırlar ve çok düşük enzim aktivite düzeyleri ölçülebilir.[2] Benzer bir yaklaşım, kütle spektrometresi kullanarak, ürüne stabil bir izotopun enkorpore olmasının veya substrattan salınmasının izlenmesidir.
En duyarlı enzim ölçümleri bir mikroskop içinden odaklanmış bir lazer kullanarak reaksiyonunu katalizleyen tek bir enzim molekülünü gözlemler.Bu ölçümler enzimin reaksiyon mekanizması sırasında kofaktörlerin floresansındaki değişimi, veya protein üzerinde belli konumlara bağlanmış floresan boyalardaki değişimi ölçer, kataliz sırasındaki hareketleri izlemek için.[3] Milyonlarca enzim molekülünün ortalama davranışını gözlemleyen geleneksel enzim kinetiğine karşın, bu tür çalışmalar tekil enzimlerin kinetiği ve dinamiği hakkında yeni bir bakış açısı sağlar.[4][5]
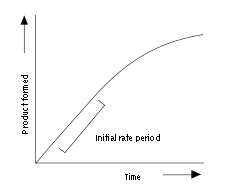
Bir enzim ölçümü için bir ilerleme eğrisi örneği yukarıda gösterilmiştir. Enzim, reaksiyonun başlamasında sonra kısa bir süre için doğrusal şekilde ürün meydana getirir. Reaksiyon ilerledikçe ve substrat tüketildikçe, hızı gittikçe azalır (substrat doyum seviyesinde olmadıkça). İlk (ve en yüksek) hızı ölçmek için, enzim ölçümleri tipik olarak substratın sadece yüzde birkaçı tüketilene kadar yapılır. Bu ilk hız döneminin uzunluğu ölçüm şartlarına bağlıdır ve milisaniyeleden saatlere kadar değişebilir. Ancak, sıvıların çok hızlı karşmasını sağlayan araçlar, bir sanıyeden kısa sürelerde ilk hızların ölçülebilmesini olanak verir.[6] Aşağıda anlatıldığı üzere, bu çok hızlı ölçümler, sabit durum öncesi (pre-steady state) kinetiğinin ölçülmesi için şarttır.
Çoğu enzim kinetik çalışmaları enzim reaksiyonlarının bu ilk, yaklaşık doğrusal, kısmı üzerine yoğunlaşır. Ancak, tüm reaksiyonu ölçüp bu verileri doğrusal olmayan bir hız denklemine uydurmak da mümkündür. Enzim reaksiyonlarının bu şekilde ölçülmesi, ilerleme eğrisi analizi (İng. progress-curve analysis) olarak adlandırılır.[7] Eğer ilk hız, hızlı kinetik ile doğru ölçülemeyecek kadar yüksek ise, bu yaklaşım yararlı bir alternatiftir.
Tek substrat reaksiyonları
Tek substrat mekanizmalı enzimlere örnek olarak, izomerazlar (triozfosfat izomeraz veya bisfosfogliserat mutaz gibi), adenilat siklaz gibi hücre içi liyazlar, ve bir RNA liyaz olan çekiçbaşı ribozimi sayılabilir.[8] Ancak, tek bir substratı olan bazı enzimler bu mekanizma kategorisi içine girmezler. Bunun bir örneği olan katalaz, substratı olan hidrojen peroksit ile reaksiyona girince yükseltgenir (okside olur) ve sonra ikinci bir substrat molekülü tarafından indirgenir. Substrat tek olsa da, modifiye olmuş bir enzim ara ürününün varlığı, katalaz mekanizmasının aslında bir pinpon (masa tenisi) mekanizması olduğunu gösterir. Bu mekanizma tipi aşağıda Çok substratlı reaksiyonlar bölümü altında açıklanmıştır.
Michaelis-Menten kinetiği

Enzimle katalizlenmiş reaksiyonlar doyumlu olduğu için, kataliz hızları artan substrata doğrusal bir tepki göstermez. Eğer ilk reaksiyon hızı çeşitli substrat konsantrasyonları ([S] ile gösterilir) için ölçülürse, reaksiyon hızı (v) [S] arttıkça artar, sağda gösterildiği gibi. Ancak, [S] yükseldikçe, enzim doygunlaşır (satüre olur) ve hız, enzimin en büyük hızı olan Vmax'a ulaşır.
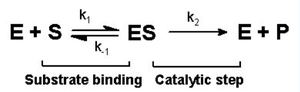
Tek substratlı reaksiyon için Michaelis-Menten kinetik modeli sağda gösterilmiştir. Enzim E ve substratı S arasında önce bir iki moleküllü reaksiyon olup bir enzim-substrat reaksiyonu ES meydana gelir. Tek moleküllü reaksiyon için enzim reaksiyonu çok karmaşık olabilirse de, tipik olarak hız belirleyici tek bir enzimatik adım vardır. Bu sayede bu reaksiyon, tek bir katalitik basamağı olan, tek moleküllü hız sabiti görünüşte kcat imiş gibi modellenebilir.

Eğer reaksiyon yolu bir veya birkaç araürün üzerinden gidiyorsa, kcat birkaç öğesel (elementer) hız sabitinin fonsiyonu olacaktır. Oysa en basit durum olan tek bir öğesel reaksiyonda (yani araürün olmaması hâlinde), öğesel tek moleküllü sabit k2'ye eşit olacaktır. Görünür tek-moleküllü hız sabiti kcat devir hızı olarak da adlandırılır ve bu, saniyedeki katalizlenen en yüksek enzim reaksiyon sayısına karşılık gelir.
Michaelis–Menten denklemi[9] (başlangıç) reaksiyon hızı v0'nin substrata bağlanma denge konumuna ve hız sabiti k2'ye nasıl bağlı olduğunu betimler.
- (Michaelis–Menten denklemi)
![{\displaystyle v_{0}={\frac {V_{\max[}{\mbox{S}}]}{K_{M}+[{\mbox{S}}]}}}](../I/m/5b95905c4ce373fddfc860731fbe53d2dce324d8.svg)
sabitler:
![{\displaystyle {\begin{aligned}K_{M}\ &{\stackrel {\mathrm {def} }{=}}\ {\frac {k_{2}+k_{-1}}{k_{1}}}\approx K_{D}\\V_{\max }\ &{\stackrel {\mathrm {def} }{=}}\ k_{cat}{[}E{]}_{tot}\end{aligned}}}](../I/m/1bea49834aba9fe3634d90051b5274c6f226ac7c.svg)
Bu Michaelis-Menten denklemi çoğu tek substratlı enzim kinetiğinin temelinde yatar. Mekanizma hakkında genel bir varsayım, araürün veya ürün inhibisyonu olmadığı, ve alosterisite veya kooperativite olmadığıdır. Bu denklemin altında yatan iki önemli varsayım daha vardır. Birincisi, sabit-durumumsuluk varsayımı (İng. quasi-steady-state assumption) (veya sahte sabit durum hipotezi) olarak adlandırılır, substrata bağlı enzim konsantrasyonundaki (ve dolayısıyla bağlı olmayan enzimin konsantrasyonundaki de) değişim hızının, ürün ve substratın konsantrasyonlarındaki değişmeden çok daha az olduğudur, dolayısıyla kompleksteki değişimin zamana bağlı değişiminin sıfıra eşitlenebileceğidir. . İkinci varsayım ise, toplam enzim konsantrayonunun zaman içinde değişmediği, yani . Bu denklemlerin türetilmesinin ayrıntıları burada bulunabilir.
![{\displaystyle d{[}ES{]}/{dt}\;{\overset {!}{=}}\;0}](../I/m/1b3e6895e4506bf8353d75418cf27091eb1ef57e.svg)
![{\displaystyle {[}E{]}_{\text{tot}}={[}E{]}+{[}ES{]}\;{\overset {!}{=}}\;{\text{const}}}](../I/m/6c707c57d7e6848dab387ad6ae0e985b847b6187.svg)
Michaelis sabiti KM'nin deneysel tanımı, enzim reaksiyon hızının Vmax'ın yarısı olduğu substrat konsantrasyonudur. Michaelis–Menten denkleminde [S] = Km substitusyonu yapılarak bu tanım doğrulanabilir, ayrıca grafik olarak da gösterilebilir. Eğer hız belirleyici enzimatik adım, substrat ayrışma hızına () kıyasla yavaş ise, Michaelis sabiti KM kabaca ES kompleksinin ayrışma katsayısı KD'ye eşittir.

Eğer , 'ye kıyasla küçükse, ve çok az ES kompleksi oluşur, yani . Dolayısıyla, ürün oluşma hızı
![{\displaystyle [S]}](../I/m/292bbb82029aa583c5d2ac5fa1d7e4fedf537d8b.svg)

![{\displaystyle [S]/(K_{M}+[S])\approx [S]/K_{M}}](../I/m/6824a6225dc4d79d295bbdb8f0517c0dd1f1f1c4.svg)
![{\displaystyle [E]_{0}\approx [E]}](../I/m/39febc25791edf75ee15e9d6b31e4e869bf46f9a.svg)
![{\displaystyle v_{0}\approx {\frac {k_{cat}}{K_{M}}}[E][S]\qquad \qquad {\text{eger }}[S]\ll K_{M}}](../I/m/b05613072c38d95b2c732735b3391504d59d2e7d.svg)
Yani, ürün oluşma hızı hem enzim konsantrasyonuna, hem de substrat konsantrasyonuna bağlıdır; psödo-ikinci derece hız sabiti olan iki moleküllü reaksiyonunun denklemine benzer bu denklem. Bu sabit, katalitik verimliliğin bir ölçütüdür. En verimli enzimler 108 - 1010 M−1 s−1 aralığında değerlerine sahiptir. Bu enzimler o kadar verimlidir ki, bir substat molekülüne her rastladıklarında bir reaksiyon katalizlerler ve dolayısıyla verimliliğin teorik üst sınırına (difüzyon sınırına) ulaşmışlardır. Bu enzimler çoğu zaman mükemmel enzim olarak adlandırılır.[10]

Michaelis-Menten denkleminin zaman bağımlı kinetik analizi için kullanımı
Michaelis-menten denklemi tarafından öngörülen reaksiyon hızlarından yararlanılarak zaman bağımlı substrat yokoluşu ve ürün oluşumu doğrudan modellenebilir. Bunu yapmak için birinci derece kimyasal kinetik denklemi içine Michaelis-Menten denklemi dahil edilir. Birinci derece kimayasal kinetiğin tarifinde Euler sayısının kullanımı ile ilgili problemin farkına varmak gerekir, yani e-k hesaplamalara sistematik bir hata sokan bir ayrık katsayıdır. Bu, tek bir katsayı kullanılarak yeniden yazılabilir, bu yeni katsayı her zaman aralığından sonraki kalan substrat miktarına karşılık gelir.[11]
![{\displaystyle [S]=[S]_{0}(1-k)^{t}\,}](../I/m/93211e467eb88a4ed3ce4b1b8a64f3645c540709.svg)
![{\displaystyle [S]=[S]_{0}(1-v/[S]_{0})^{t}\,}](../I/m/fc767ed4ec3fb17dbb2b342b438ca22f3a0c5e15.svg)
![{\displaystyle [S]=[S]_{0}(1-(V_{\max[}S]_{0}/(K_{M}+[S]_{0})/[S]_{0}))^{t}\,}](../I/m/d29e711bf38226423c5d45b6d47738477796d98f.svg)
Michaelis–Menten denklemi için doğrusal grafikler
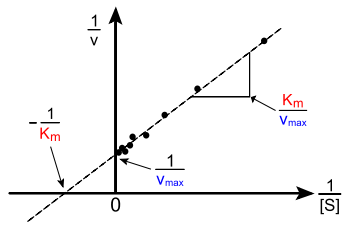
Yukarıda gösterilen, [S]'ye bağlı v grafiği doğrusal değildir; grafik, düşük [S] için başlangıçta doğrusal olsa dahi, yüksek [S]'de eğilerek doyuma ulaşır. Bu konuda yapılan ilk çalışmalarda, bu nonlineerlik KM ve Vmax'ın hatasız olarak kestirilmesini zor kılmaktaydı. Bu yüzden, bazı araştırmacılar Michaelis-Menten denkleminin lineerleştiren grafik yöntemler geliştirdiler: Lineweaver–Burk grafiği, Eadie–Hofstee çizimi ve Hanes–Woolf grafiği gibi. Bu lineer gösterimlerin hepsi verinin görüntülenmesi için yararlıdır, ama kinetik parametrelerinin belirlenmesi için günümüzde bilgisayar yazılımları mevcuttur ve nonlineer regresyon yöntemleri kullanarak daha hatasız sonuçlar elde edilmesi mümkündür.[12]
Lineweaver-Burk grafiği veya çifte evrik (İng. reciprocal) grafiği kinetik veriyi görüntülemek için yaygın kullanılır. Bunu elde etemk için Michaelis-Menten denkleminin iki tarafının evrik yapılır. Sağda gösterildiği gibi, bu, Michaelis-Menten denkleminin doğrusal bir biçimidir ve y = mx + c denklemli bir doğru üretir, y ekseni kesişim noktası 1/Vmax'ye x eksen kesişimi -1/KM'ye karşılık gelir.
![{\displaystyle {\frac {1}{v}}={\frac {K_{M}}{V_{\max[}{\mbox{S}}]}}+{\frac {1}{V_{\max }}}}](../I/m/3dc9af4681099fefe2aa5a0bf39e9f837bb894d4.svg)
Tabii ki, negatif 1/[S] değerleri için deneysel değerler elde edilemez; 1/[S] = 0 alt sınırı (y kesişimi) sonsuz substrat konsantrasyonuna karşılık gelir, orada 1/v=1/Vmax'dır, sağda gösterildiği gibi. Dolayısıyla x-kesişimi pozitif konsantrasyonlarda elde edilen deneysel verilerin bir ekstrapolasyonudur. Daha genel olarak, Lineweaver–Burk grafiği düşük konsantrasyonlarda elde edilen ölçümlerin önemini çarpıtır ve dolayısıyla Vmax ve KM'nin hatalı kestirilmesine yol açabilir.[13] Daha hatasız bir lineer grafikleme yöntemi Eadie-Hofstee grafiğidir. Bu durumda v, v/[S]'ye bağlı olarak grafiklenir. Yaygın kullanılan yöntemlerden üçüncüsü olan Hanes-Woolf grafiğinde, [S]/v, [S]'ye bağlı olarak grafiklenir. Genelde, data normalizasyonu deneysel iş miktarını azaltmaya yarar, sonucun güvenilirliğini artırabilir ve hem grafik hem de sayısal analiz için uygundur.[14]
Kinetik sabitlerin pratik anlamı
Enizm kinetiğinin araştırılması iki temel nedenden dolayı önemlidir. Birincisi, enzimlerin nasıl çalıştığının anlaşılmasını sağlar, ikincisi, enzimlerin organizmalarda nasıl davrandığını öngörmeye yardımcı olur. Yukarıda tanımlanan kinetik katsayılar KM ve Vmax, metabolima kontrolünde enzimlerin nasıl çalıştığının anlaşılmasında önemli rol oynar.
Bu öngörülerin yapılması sıradan değildir, basit sistemler için dahi. Örneğin, oksaloasetat, mitokondride bulunan malat dehidrojenaz tarafından meydana gelir. Oksaloasetat sonra sitrat sentaz, fosfoenolpirüvat karboksikinaz veya aspartat aminotransferaz tarafından tüketilebilir ve, sırasıyla, sitrik asit döngüsü, glükoneojenez veya aspartik asit biyosentezini besleyebilir. Bu yolaklardan her birine ne kadar oksaloasetat gittiğini öngörmek için oksaolasetat konsantrasyonunu bilmek, ayrıca bu enzimlerin konsantrasyon ve kinetik özelliklerini bilmek gerekir. Metabolik yolakların davranışlarını öngörüsünün en karmaşık kısmı, tüm bir organizmaya ait matematiksel modellerin içine girmesi gereken kinetik ve gen ifadesi verilerinin sentezini yapılmasındadır. Buna alternatif olarak, kinetik metabolizma probleminin faydalı bir sadeleştirmesi, enzim kinetiklerini dikkate almayıp sadece reaksiyon ağının stokyometresi hakkındaki bilgileri dayanmaktır, bu yöntem akı denge analizi olarak adlandırılır.[15][16].
Ararürünlü Micahelis-Menten kinetiği
Basit olan şu durum ile başlayalım:

burada bir enzim ve araürün mevcuttur, araürün ikinci adımda ürüne dönüşür. Bu durumda çok benzer bir denklem ortaya çıkar.[17]
![{\displaystyle {\begin{aligned}v_{0}&=k_{cat}{\frac {{[}S{]}{[}E{]}_{0}}{K_{M}^{\prime }+{[}S{]}}}\end{aligned}}}](../I/m/8d9f41e22b3bc50a588ef17c9fdd38434bb18c2e.svg)
ama sabitler farklıdır

Sınırlayıcı durum olan için, yani EI 'den E + P'ye olan adım bir evvelki adımdan çok daha hızlı olunca, ilk denklemi elde edilir. Matematik olarak, ve .



Çok-substratlı reaksiyonlar
Çok substratlı reaksiyonlarda subsratların nasıl bağlandığını ve hangi sırayla bağlandığını belirten karmaşık hız denklemleri vardır. Eğer A substratının konsantrasyonu sabit tutulur ve B substratınınki değiştirilirse, bu reaksiyonların analizi çok basitleşir. Bu şartlar altında, enzim tek substratlı bir enzim gibi davranış gösterir, v'yi [S]'ye bağlı olarak grafiklendiğinde, B substatı için görünür KM ve Vmax sabitleri elde edilir. Eğer bu ölçümler A'nın farklı sabit konsantrasyonları için yapılırsa, bu veriler kullanılarak reaksiyonun mekanizması anlaşılabilir. İki substrat A ve B'yi alıp bunları iki ürün P ve Q'ya dönüştüren bir enzim için iki tip mekanizma vardır, üçlü kompleks veya pinpon.
Üçlü kompleks mekanizmaları
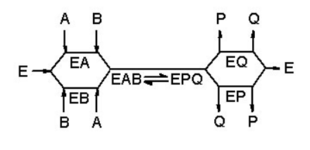
Bu enzimlerde, her iki substrat enzime aynı anda bağlanıp bir EAB üçlü kompleksi meydana getirir. Bağlanma sırası rastgele olabilir (rastgele mekanizmada) veya substratların belli bir sıra sıra ile bağlanması gerekebilir (sıralı mekanizmada). Üçlü kompleks mekanizmalı bir enzim için bir v-[S] eğriler kümesi (sabit A, değişen B) bir Lineweaver-Burk grafiğinde çizilirse, doğrular bir noktada kesişir.
Üçlü kompleks mekanizmalı enzimler arasında glutatyon S-transferaz,[18] dihidrofolat reductaz[19] and DNA polymeraz sayılabilir.[20] Dihidrofoloat redüktaz ve DNA polimeraz için üçlü-kompleks mekanizmalarını gösteren kısa animasyonlar şu bağlantıda izlenebilir.{{Ref_label|B|β|none}[γ]
Pinpon mekanizması
Sağda gösterildiği gibi, pinpon mekanizmalı enzimler iki durumda bulunabilir: asıl hâlleri olan E ve kimyasal olarak modifiye bir hâl olan E*. Enzimin bu modifiye olmuş hâli "reaktif araürün" olarak adlandırılır. Bu mekanizmada, substrat A enzime bağlanır, aktif bölgeye bir kimyasal grup aktararak, enzimi E*'a dönüştürür, ve sonra enzimden salınır. İlk substrat salındıktan sonra ancak substrat B bağlanabilir ve modifiye olmuş enzimle reaksiyona girip modifiye olmamış E biçimini yeniden oluşturur. Pinpon mekanizmalı bir enzim için bir v-[S] eğriler kümesi (sabit A, değişen B), Lineweaver-Burk grafiğinde çizilirse, doğrular bir noktada kesişir. Bu grafik, ikincil grafik olarak adlandırılır.
Pinpon mekanizmalı enzimler arasında bazı oksidoredüktazlar (tiyoredoksin peroksidaz[21] gibi), transferazlar (asilnöraminat citidililtransferaz[22] gibi) ve serin proteazlar (tripsin ve kimotripsin gibi[23]) sayılabilir. Serin proteazlar yaygın ve çeşitli üyelere sahib bir enzim ailesidir, aralarında sindirici enzimler (tripsin, kimotripsin ve elastaz), kan pıhtılaşma kaskadının bazı enzimleri ve diğerleri sayılabilir. Serin proteazlarda, E* araürünü bir asil-enzim türüdür, aktif bölge serin kalınıtısının bir protein substratının peptit bağına saldırısı sonucu meydana gelir. Kemotripsin mekanizmasını gösteren bir kısa animasyona bir bağlantı şurda verilmiştir[δ]
Michaelis–Menten dışı kinetikler
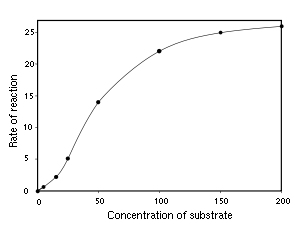
Bazı enzimlerin v - [S] grafiği sigmoid şeklindedir, bu şekilli grafik, substratın aktif bölgeye bağlanmasında kooperatif bağlanma olduğunun belirtisidir. Bu demektir ki, bir substratın bağlanması sonraki substrat moleküllerinin bağlanmasına etki eder. Bu davranış, birbiriyle etkleşen birkaç aktif bölgenin olduğu mültimerik proteinlerde yaygındır.[24] Kooperasyon mekanizması, hemoglobinde görülen bağlanma sürecine benzer, bir substratın aktif bölgeye bağlanması, diğer aktif bölgelerin substrat moleküllere olan afinitesini değiştirir. Pozitif kooperatiflik durumunda, ilk substratın bağlanması sonucu diğer substratlarına olan afinite (ilgi) artar. Negatif kooperatiflikte ise, ilk substratın bağlanması, diğer substratlara olan afinitenin azalmasına neden olur.
Allosterik enzimlere örnek olarak memeli tirozil tRNA-sentetaz (negatif kooperatiflik gösterir)[25], bakteryel aspartat transkarbamoylaz[26] ve fosfofructokinaz (pozitif kooperatiflik gösterir)[27] sayılabilir.
Kooperatiflik oldukça yaygındır ve substrat konsantrasyonundaki değişikliğe bağlı olarak enzimlerin tepkilerinin düzenlemeye yarar. Pozitif kooperatiflik enzimleri [S]'ye daha duyarlı kılar ve dar bir konsantrasyon aralığında enzim aktivitesinin büyük değişklik göstermesini sağlar. Buna karşın, negatif kooperatiflik enzimleri [S]'de küçük değişkliklere az duyarlı kılar.
Hill denklemi[28] Michaelis*Menten kinetiğine uymayan sistemlerdeki kooperatiflik derecesini nicel olarak betimlemek için kullanılır. Türetilmiş Hill katsayısı n substratın bir aktif bölgeye bağlanmasının substratın diğer aktif bölgelere bağlanmasının ne kadar etkilediğinin ölçütüdür. Hill kasayısı <1 olması negatik kooperatiflik, Hill katsayısı >1 olması pozitif kooperatifliği belirtir.
Sabit hâl öncesi kinetik
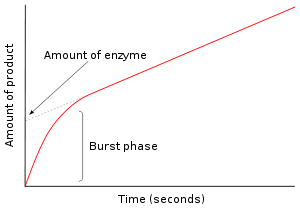
Bir enzim substratı ile karıştırlmasından sonraki ilk anda henüz bir ürün oluşmamış ve araürün yoktur. Bunu takip eden milisaniylerin araştırması sabit hâl öncesi kinetik veya çoğuşma kinetiği (İngilizce Burst kinetics) olarak adlandırılır. Dolayısıyla, sabit hâl öncesi kinetiği, sabit hâl konsantrasyolarına ulaşılana dek enzim substrat araürünlerinin (ES veya E* gibi) oluşumu ve tükenişi ile ilgilidir.
Bu yaklaşım ilk defa kimotripsin tarafından katalizlenen hidroliz reaksiyonuna uygulanmıştır.[29] Genelde, bir araürünün tespiti, bir enzimin takip ettiği mekanizmayı araştırmada çok önemli bir delildir. Örneğin, yukarıda gbelirtilen pinpon mekanizmasında, hızlı kinetik ölçümler bir P ürününün enzimden salınmasını ve değişime uğramış bir E* enzim araürününün oluşumu izlenebilir.[30] Kimotripsin durumunda, aktif bölgedeki nükleofilik serin substrata saldırarak bir asil-enzim araürünü meydana gelir.
Sağdaki resimde, reaksiyonun ilk birkaç saiyesi sırasında enzim hızla E* oluşturur. Sonra, sabit hâle yaklaştıkça reaksiyon yavaşlar. Reaksiyonun bu hızlı "çoğuşma" evresi, enzimdeki tek bir devinime karşılık gelir. Dolayısıyla, bu patlama sırasında salınan ürün miktarı (grafikte y eksenindeki kesim noktasına karşılık gelir), aynı zamanda reaksiyondaki fonksiyonel enzim miktarına eşittir.[31]
Kimyasal mekanizma
Enzim kinetiğinin ölçümünde önemli bir amaç, enzim reaksiyonunun kimyasal mekanizmasının, yani substratın ürüne dönüştürülmesindeki kimyasal adımlar dizisinin belirlenmesidir. Yukarıda anlatılan bu kinetik yaklaşım ile araürünlerin ne hızla oluştuğu ve birbirine dönüştüğünü gösterir ama bu araürünlerin tam olarak ne olduğunu tespit edemez.
Farklı çözelti şartlarında yapılan veya biraz modifiye edilmiş enzim veya substratlar üzerinde yapılan kinetik ölçümler çoğu zaman bu kimyasal mekanizmanın anlaşılmasına yardımcı olur, çünkü hız belirleyici adım veya reaksiyon araürünlerini ortaya çıkarır. Örneğin, bir hidrojen atomuna olan bir kovalent bağın kırılması sık görülen bir hız belirleyici adımdır. Olasıl hidrojen transferlerinden hangisinin hız belirleyici olduğu, her bir hidrojeni (onun kararlı izotopu olan) döteryum ile değiştirmenin kinetik etkilerini ölçülerek gösterilebilir. Kinetik izotop etkisinden dolayı döteryum bağlarının kırılması hidrojene olan kovalent bağların kırılmasından daha zordur, bu yüzden kritik hidrojen değiştirildiğinde reaksiyon hızı değişir.[32] Başka izotop substitusyonları ile de (13C/12C ve 18O/16O) benzer etkilerin ölçülmesi mümkündür, ama bu etkiler aynı derecede bariz değildir.[33]
İzotoplar substrat molekülünün farklı kısımlarının kaderini ortaya çıkarmak için de kullanılabilir. Örneğin, bazen nihai üründeki bir oksijenin kaynağını ayırt etmek zor olabilir, çünkü bu atom sudan veya substrattan geliyor olabilir. Hangisi olduğunu belirlemek için, reaksiyona katılan çeşitli moleküllere oksijenin kararlı izotopu olan 18O sistematik olarak substitüe edilir ve üründe bu izotopun varlığına bakılır.[34] Kimyasal mekanizmanın anlaşılması için kullanılabilen diğer yöntemler, kinetik ve izotop etkileri farklı pH şartlarında,[35] enzime bağlanan metal iyonları veya diğer kofaktörleri değiştirerek,[36] korunmuş amino asit kalıntılarının mutagenezi ile veya substrat analoglarının varlığında enzimin davranıçını incelemektir.[37]
Enzim inhibisyonu ve aktivasyonu
Enzim inhibitörleri enzim aktivitesini azaltan veya ortadan kaldıran mpoleküllerdir, buna karşın enzim aktivatörleri enzimlerin katalitik aktivitesini artıran moleküllerdir. Bu etkileşimler ya tersinir (yani, inhibitörünün giderilmesi enzim aktivitesini geri getirir) veya tersinmez olabilir (yani inhibitör temelli olarak enzimi inaktive eder).
Tersinir inhibitörler
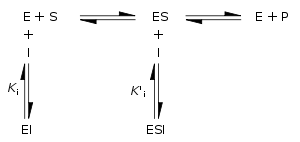
Geleneksel olarak, tersinir enzim inhibitörleri Km ve Vmax üzerindeki etkilerine bağlı olarak yarışmalı (İngilizce competitive), sınırlı yarışmalı (non-competitive), yarışmasız (uncompetitive) veya karışık inhibisyon olarak sınıflandırılmışlardır. Bu farklı etkiler inhibitörün ya E enzimine, ya ES enzim-substrat kompleksine, ya da her ikisine birden bağlanmasının sonucudur, sağdaki şekil ve aşağıdaki tabloda gösterildiği üzere. İnhibitörün tipine ayırt etmek için, inhibitör konsantrasyonuna bağlı olarak enzim kinetiği incelenir. Bu dört tip inhibitör, inhibitör konsantrasyonuna bağlı olarak farklı Lineweaver–Burke ve Eadie–Hofstee grafikleri oluşturur.[13] Sadelik için ilgili denklemlerde şu iki sembol kullanılır:
- and
![{\displaystyle \alpha =1+{\frac {[{\mbox{I}}]}{K_{i}}}}](../I/m/7d05bffbc19ef9047581c87887ae0eea5c11e7fe.svg)
![{\displaystyle \alpha ^{\prime }=1+{\frac {[{\mbox{I}}]}{K_{i}^{\prime }}}}](../I/m/e8b2033b5725a8b85d7425d409192cf11885c199.svg)
Burada Ki ve K'i, sırasıyla, enzime ve enzim-substrat kompleksinin ayrışma sabitleridir. Tersinir bir inhibitörün varlığında, enzimin görünen Km ve Vmax değerleri, sırasıyla, (α/α')Km ve (1/α')Vmax olur,aşağıdaki tipik durumlar için gösgterildiği gibi.
| İnhibisyon tipi | Görünür Km | Görünür Vmax | ||
| Ki sadece | () | yarışmalı | ||
| Ki' sadece | () | yarışmasız | ||
| Ki = Ki' | () | sınırlı yarışmalı | ||
| Ki ≠ Ki' | () | karışık |










non-lineer regresyon ile enzim kinetik verilerini hız denklemlerine uydurunca[38] ayrışma katsayıları Ki ve K'i için yüksek doğruluklu olarak kestirmek mümkün olur.
Denklemlerin basit bir düzenlemesi inhibitör bağlanması ve maksimum hız arasındaki ilişkiyi ortaya koyar.
![{\displaystyle {\cfrac {V_{\max }}{1+{\cfrac {[I]}{K_{i}}}}}}](../I/m/77b235af32e059b205f8205c562158c86988b340.svg)
![{\displaystyle {\cfrac {V_{\max }}{\cfrac {[I]+K_{i}}{K_{i}}}}}](../I/m/033f13695d07302c106ac09cbafc690d81f5a5b6.svg)
Aşağıya sıfır ekleyince ([I]-[I])
![{\displaystyle {\cfrac {V_{\max }}{\cfrac {[I]+K_{i}}{[I]+K_{i}-[I]}}}}](../I/m/42e34b0927bf9484b8a664e022d3fd6ba0ad2326.svg)
[I]+Ki ile bölünce
![{\displaystyle {\cfrac {V_{\max }}{\cfrac {1}{1-{\cfrac {[I]}{[I]+K_{i}}}}}}}](../I/m/b3c1e590a1ee697acaebbee65eea01ff1acdc12f.svg)
![{\displaystyle V_{\max }-V_{\max }{\cfrac {[I]}{[I]+K_{i}}}}](../I/m/8c279152b9fce0341d0b79b769c617c038da50c5.svg)
Yani, reaksiyon hızının substrat ile etkileşen enzim oranına bağlı olduğu Michaelis–Menten denklemine benzer şekilde,
substrata bağlı enzim oranı
![{\displaystyle {\cfrac {[S]}{[S]+K_{m}}}}](../I/m/d4864d15c155b606d4e3c9e6318eda5ba0e38b38.svg)
inhibitöre bağlı enzim oranı
![{\displaystyle {\cfrac {[I]}{[I]+K_{i}}}}](../I/m/a1c9c3a9de93daeb179488d6523b7ea9a96e8cba.svg)
İnhibitörün etkisi, inhibitörle etkileşen enzim popülasyonunun yüzdesinin bir sonucudur. Bu denklemle ilgili tek sorun, inhbitör bağlanması ile enzimin mutlak olarak inhibe olduğunu varsaymasıdır, oysa bu etki substrat kullanımının %100 ila %1'in biraz fazlası arasında değişebilir. Değişken dereceli inhibisyonu hesaba katmak için denkleme bir delta Vmax terimi eklenir.
![{\displaystyle V_{\max }-\Delta V_{\max }{\cfrac {[I]}{[I]+K_{i}}}}](../I/m/90f5601fefd8114c165ac3dfb739e0642e62610c.svg)
veya
![{\displaystyle V_{\max }1-(V_{\max }1-V_{\max }2){\cfrac {[I]}{[I]+K_{i}}}}](../I/m/eebe96aec4c5c5472dde69ce46cf75ed805676fb.svg)
Bu terim sayesinde, inhibitör popülasyondaki enzim molekülleri ile etkileşirken kalan enzim aktivitesi tanımlananbilir. Ayrıca, bu terimin denkleme dahil edilmesinin bir avantajı, ikincil Vmax terimi ilk terimden daha büyük olması halinde aktivasyon olasılığının da hesaplanmasını mümkün kılmasıdır. Aktivasyon olasılığını hesaba katmak için, İnhibitör için kullanılan "I" harfi yerine, aşağıda "X" olarak gösterilen bir modifikatör kavramına geçilebilir.
![{\displaystyle V_{\max }1-(V_{\max }1-V_{\max }2){\cfrac {[X]}{[X]+K_{x}}}}](../I/m/9d29eb761d8d80d1b27749d77cc921c022ce1fa7.svg)
Bu terminoloji Michaelis-Menten denklemineki maksimal hız ile ilşkili kinetik etkileri daha basit bir şekilde ele almaya sağlamasına karşın, Km ile ilişkili etkileri tarif etmekte ortaya çıkabilen potansiyel problemleri de ortaya çıkarır. Enzimin substratına afinitesi ile ilgili olan Km, çoğu durumda enzim-inhbitör etkileşiminin sonucu olarak enzimin bağlanma yerindeki potansiyel değişikliklerle ilişkili olmalıdır. Bu yüzden, Vmax'yi modüle eden, yukarıdaki benzer bir terim olması çoğu durum için uygun olurdu:[39]
![{\displaystyle K_{m}1-(K_{m}1-K_{m}2){\cfrac {[X]}{[X]+K_{x}}}}](../I/m/02d4b89078d0ef0508d2a49553de50dac82e43fe.svg)
Tersinmez inhibitörler
Enzim inhibitörleri enzimleri tersinmez olarak da inaktive edebilirler, genelde aktif bölgedeki amino asit kalıntısını değişime uğratarak. Kendilerini de yok ettikeri için "intihar substratı" olarak düşünülebilecek olan bu inhibitörler, reaksiyon sırasında üssel bir azalma gösterirler ve konsantrasyona bağlı olarak enzime olan etkileri genelde doyum özelliği gösterir. Doyum noktasının altında bu reaksiyonlar inhibitör için birinci derece reaksiyon kinetiği gösterirler.
Kataliz mekanizması
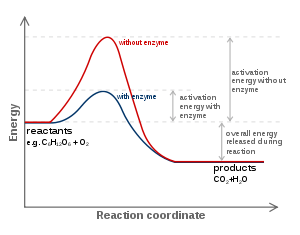
Enzim-substrat etkileşimi ile ilgili favori model, indüklenmiş uyum modelidir.[40] Bu modele göre, enzim ile substrat arasındaki ilk etkileşimin zayıftır, ancak bu zayıf etkileşimler enzimde konformasyonel değişiklikler doğurur (indükler), ve bunun sonucu olarak bağlanma güçlenir. Bu konformasyonel değişiklikler ayrıca aktif bölgedeki katalitik amino asit kalıntılarını, substratta reaksiyon sonucu değişecek kimyasal bağların yakınına getirir.[41] Konformasyonel değişimler dairesel dikroizm veya eşlek polarizasyon enterformetrisi (İng. dual polarisation interferometry) ile ölçülebilir. Bağlanma olduktan sonra, kataliz mekanizmaları sayesinde, reaksiyon için alternatif kimyasal yolaklar sağlanarak reaksiyonun geçiş halinin enerjisi alçalır. Bunun olmasını sağlayan kataliz mekanizmaları arasında: bağ gerginliği yoluyla kataliz; yakınlık ve doğrultu ile kataliz; aktif bölge proton vericileri veya alıcıları ile kataliz; kovalent kataliz; ve kuvantum tünellemesi sayılabilir.[30][42]
Enzim kinetiği ölçümleri bir enzim tarafından hangi kataliz yolunun kullanıldığını kanıtlayamaz. Ancak, bazı kinetik veriler, başka yöntemlerle doğrulanabilecek olasılıkları öne sürebilir. Örneğin, sabit-hâl öncesi çoğuşmalı pinpon kinetiği gösteren bir reaksiyon, kovalent katalizin bu enzim reaksiyonunda önemli olabileceğini ima eder. Alternatif olarak, Vmax üzerinde kuvvetli bir pH etkisi olması ama Km üzerinde bir pH etksisi olmaması, katalizin olabilmesi için aktif bölgedeki bir amino asit kalıntısının belli bir iyonizasyon hâlinde olması gerektiğine işaret edebilir.
Ayrıca bakınız
- Protein dinamikleri
Dipnotlar
α. ^ Interaktif Michaelis–Menten kinetiği eğitseli (Java gereklidir)
β. ^ dihidrofolat redüktaz mekanizması (Gif)
γ. ^ DNA polimeraz mekanizması (Gif)
δ. ^ Şimotripsin mekanziması (Flash gereklidir)
Kaynaklar
- ↑ Wrighton, Mark S.; Ebbing, Darrell D. (1993). General chemistry (4th bas.). Boston: Houghton Mifflin. ISBN 0-395-63696-5.
- ↑ Danson, Michael; Eisenthal, Robert (2002). Enzyme assays: a practical approach. Oxford [Oxfordshire]: Oxford University Press. ISBN 0-19-963820-9.
- ↑ Xie XS, Lu HP (June 1999). "Single-molecule enzymology". J. Biol. Chem. 274 (23): 15967–70. DOI:10.1074/jbc.274.23.15967. PMID 10347141. http://www.jbc.org/cgi/content/full/274/23/15967.
- ↑ Lu H (2004). "Single-molecule spectroscopy studies of conformational change dynamics in enzymatic reactions". Current pharmaceutical biotechnology 5 (3): 261–9. DOI:10.2174/1389201043376887. PMID 15180547.
- ↑ Schnell J, Dyson H, Wright P (2004). "Structure, dynamics, and catalytic function of dihydrofolate reductase". Annual review of biophysics and biomolecular structure 33: 119–40. DOI:10.1146/annurev.biophys.33.110502.133613. PMID 15139807.
- ↑ Gibson QH (1969). "Rapid mixing: Stopped flow". Methods Enzymol 16: 187–228. DOI:10.1016/S0076-6879(69)16009-7.
- ↑ Duggleby RG (1995). "Analysis of enzyme progress curves by non-linear regression". Methods Enzymol 249: 61–90. DOI:10.1016/0076-6879(95)49031-0. PMID 7791628.
- ↑ Murray JB, Dunham CM, Scott WG (January 2002). "A pH-dependent conformational change, rather than the chemical step, appears to be rate-limiting in the hammerhead ribozyme cleavage reaction". J. Mol. Biol. 315 (2): 121–30. DOI:10.1006/jmbi.2001.5145. PMID 11779233.
- ↑ Michaelis L. and Menten M.L. Kinetik der Invertinwirkung Biochem. Z. 1913; 49:333–369 İngilizce çevirisi Accessed 6 April 2007
- ↑ Stroppolo ME, Falconi M, Caccuri AM, Desideri A (2001). "Superefficient enzymes". Cell. Mol. Life Sci. 58 (10): 1451–60. DOI:10.1007/PL00000788. PMID 11693526.
- ↑ Walsh R, Martin E, Darvesh S. A method to describe enzyme-catalyzed reactions by combining steady state and time course enzyme kinetic parameters. Biochim Biophys Acta. 2010 Jan;1800:1-5
- ↑ Jones ME (1 January 1992). "Analysis of algebraic weighted least-squares estimators for enzyme parameters". Biochem. J. 288 (Pt 2): 533–8. PMC 1132043. PMID 1463456. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1132043.
- 1 2 Tseng SJ, Hsu JP (August 1990). "A comparison of the parameter estimating procedures for the Michaelis-Menten model". J. Theor. Biol. 145 (4): 457–64. DOI:10.1016/S0022-5193(05)80481-3. PMID 2246896.
- ↑ Bravo IG, Busto F, De Arriaga D, et al. (September 2001). "A normalized plot as a novel and time-saving tool in complex enzyme kinetic analysis". Biochem. J. 358 (Pt 3): 573–83. PMC 1222113. PMID 11577687. http://www.biochemj.org/bj/358/0573/bj3580573.htm.
- ↑ Almaas E, Kovács B, Vicsek T, Oltvai ZN, Barabási AL (February 2004). "Global organization of metabolic fluxes in the bacterium Escherichia coli". Nature 427 (6977): 839–43. DOI:10.1038/nature02289. PMID 14985762.
- ↑ Reed JL, Vo TD, Schilling CH, Palsson BO (2003). "An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR)". Genome Biol. 4 (9): R54. DOI:10.1186/gb-2003-4-9-r54. PMC 193654. PMID 12952533. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=193654.
- ↑ Denklemin türetilmesi için bakınız: here
- ↑ Dirr H, Reinemer P, Huber R (March 1994). "X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function". Eur. J. Biochem. 220 (3): 645–61. DOI:10.1111/j.1432-1033.1994.tb18666.x. PMID 8143720.
- ↑ Stone SR, Morrison JF (July 1988). "Dihydrofolate reductase from Escherichia coli: the kinetic mechanism with NADPH and reduced acetylpyridine adenine dinucleotide phosphate as substrates". Biochemistry 27 (15): 5493–9. DOI:10.1021/bi00415a016. PMID 3052577.
- ↑ Fisher PA (1994). "Enzymologic mechanism of replicative DNA polymerases in higher eukaryotes". Prog. Nucleic Acid Res. Mol. Biol. 47: 371–97. DOI:10.1016/S0079-6603(08)60257-3. PMID 8016325.
- ↑ Akerman SE, Müller S (August 2003). "2-Cys peroxiredoxin PfTrx-Px1 is involved in the antioxidant defence of Plasmodium falciparum". Mol. Biochem. Parasitol. 130 (2): 75–81. DOI:10.1016/S0166-6851(03)00161-0. PMID 12946843. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T29-49321S6-2&_coverDate=08%2F31%2F2003&_alid=466052639&_rdoc=1&_fmt=&_orig=search&_qd=1&_cdi=4913&_sort=d&view=c&_acct=C000050221&_version=1&_urlVersion=0&_userid=10&md5=965e18a4c2ad0af711ba05d1a46dc855.
- ↑ Bravo IG, Barrallo S, Ferrero MA, Rodríguez-Aparicio LB, Martínez-Blanco H, Reglero A (September 2001). "Kinetic properties of the acylneuraminate cytidylyltransferase from Pasteurella haemolytica A2". Biochem. J. 358 (Pt 3): 585–98. PMC 1222114. PMID 11577688. http://www.biochemj.org/bj/358/0585/bj3580585.htm.
- ↑ Kraut J (1977). "Serine proteases: structure and mechanism of catalysis". Annu. Rev. Biochem. 46: 331–58. DOI:10.1146/annurev.bi.46.070177.001555. PMID 332063. http://arjournals.annualreviews.org/doi/abs/10.1146/annurev.bi.46.070177.001555?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dncbi.nlm.nih.gov.
- ↑ Ricard J, Cornish-Bowden A (July 1987). "Co-operative and allosteric enzymes: 20 years on". Eur. J. Biochem. 166 (2): 255–72. DOI:10.1111/j.1432-1033.1987.tb13510.x. PMID 3301336.
- ↑ Ward WH, Fersht AR (July 1988). "Tyrosyl-tRNA synthetase acts as an asymmetric dimer in charging tRNA. A rationale for half-of-the-sites activity". Biochemistry 27 (15): 5525–30. DOI:10.1021/bi00415a021. PMID 3179266.
- ↑ Helmstaedt K, Krappmann S, Braus GH (September 2001). "Allosteric regulation of catalytic activity: Escherichia coli aspartate transcarbamoylase versus yeast chorismate mutase". Microbiol. Mol. Biol. Rev. 65 (3): 404–21, table of contents. DOI:10.1128/MMBR.65.3.404-421.2001. PMC 99034. PMID 11528003. http://mmbr.asm.org/cgi/content/full/65/3/404.
- ↑ Schirmer T, Evans PR (January 1990). "Structural basis of the allosteric behaviour of phosphofructokinase". Nature 343 (6254): 140–5. DOI:10.1038/343140a0. PMID 2136935.
- ↑ Hill, A. V. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. (Lond.), 1910 40, iv-vii.
- ↑ Hartley BS, Kilby BA (February 1954). "The reaction of p-nitrophenyl esters with chymotrypsin and insulin". Biochem. J. 56 (2): 288–97. PMC 1269615. PMID 13140189. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1269615.
- 1 2 Fersht, Alan (1999). Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. San Francisco: W.H. Freeman. ISBN 0-7167-3268-8.
- ↑ Bender ML, Begué-Cantón ML, Blakeley RL, et al. (December 1966). "The determination of the concentration of hydrolytic enzyme solutions: alpha-chymotrypsin, trypsin, papain, elastase, subtilisin, and acetylcholinesterase". J. Am. Chem. Soc. 88 (24): 5890–913. DOI:10.1021/ja00976a034. PMID 5980876.
- ↑ Cleland WW (January 2005). "The use of isotope effects to determine enzyme mechanisms". Arch. Biochem. Biophys. 433 (1): 2–12. DOI:10.1016/j.abb.2004.08.027. PMID 15581561. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6WB5-4DD8DXJ-8&_coverDate=01%2F01%2F2005&_alid=466049795&_rdoc=1&_fmt=&_orig=search&_qd=1&_cdi=6701&_sort=d&view=c&_acct=C000050221&_version=1&_urlVersion=0&_userid=10&md5=cf322c4a1c7db6b9f89551a8469a1a2d.
- ↑ Northrop D (1981). "The expression of isotope effects on enzyme-catalyzed reactions". Annu. Rev. Biochem. 50: 103–31. DOI:10.1146/annurev.bi.50.070181.000535. PMID 7023356.
- ↑ Baillie T, Rettenmeier A (1986). "Drug biotransformation: mechanistic studies with stable isotopes". Journal of clinical pharmacology 26 (6): 448–51. PMID 3734135.
- ↑ Cleland WW (1982). "Use of isotope effects to elucidate enzyme mechanisms". CRC Crit. Rev. Biochem. 13 (4): 385–428. DOI:10.3109/10409238209108715. PMID 6759038.
- ↑ Christianson DW, Cox JD (1999). "Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes". Annu. Rev. Biochem. 68: 33–57. DOI:10.1146/annurev.biochem.68.1.33. PMID 10872443.
- ↑ Kraut D, Carroll K, Herschlag D (2003). "Challenges in enzyme mechanism and energetics". Annu. Rev. Biochem. 72: 517–71. DOI:10.1146/annurev.biochem.72.121801.161617. PMID 12704087.
- ↑ Leatherbarrow RJ (December 1990). "Using linear and non-linear regression to fit biochemical data". Trends Biochem. Sci. 15 (12): 455–8. DOI:10.1016/0968-0004(90)90295-M. PMID 2077683.
- ↑ Walsh R, Martin E, Darvesh S. A versatile equation to describe reversible enzyme inhibition and activation kinetics: modeling beta-galactosidase and butyrylcholinesterase. Biochim Biophys Acta. 2007 1770:733-46.
- ↑ Koshland DE (February 1958). "Application of a Theory of Enzyme Specificity to Protein Synthesis". Proc. Natl. Acad. Sci. U.S.A. 44 (2): 98–104. DOI:10.1073/pnas.44.2.98. PMC 335371. PMID 16590179. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=335371.
- ↑ Hammes G (2002). "Multiple conformational changes in enzyme catalysis". Biochemistry 41 (26): 8221–8. DOI:10.1021/bi0260839. PMID 12081470.
- ↑ Sutcliffe M, Scrutton N (2002). "A new conceptual framework for enzyme catalysis. Hydrogen tunnelling coupled to enzyme dynamics in flavoprotein and quinoprotein enzymes". Eur. J. Biochem. 269 (13): 3096–102. DOI:10.1046/j.1432-1033.2002.03020.x. PMID 12084049. http://content.febsjournal.org/cgi/content/full/269/13/3096.
Daha çok okuma için
Giriş düzeyi
- Cornish-Bowden, Athel (2004). Fundamentals of enzyme kinetics (3rd bas.). London: Portland Press. ISBN 1-85578-158-1. (İngilizce)
- Stevens, Lewis; Price, Nicholas C. (1999). Fundamentals of enzymology: the cell and molecular biology of catalytic proteins. Oxford [Oxfordshire]: Oxford University Press. ISBN 0-19-850229-X. (İngilizce)
- Bugg, Tim (2004). Introduction to Enzyme and Coenzyme Chemistry. Cambridge, MA: Blackwell Publishers. ISBN 1-4051-1452-5. (İngilizce)
İleri düzey
- Segel, Irwin H. (1993). Enzyme kinetics: behavior and analysis of rapid equilibrium and steady state enzyme systems (New bas.). New York: Wiley. ISBN 0-471-30309-7. (İngilizce)
- Fersht, Alan (1999). Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. San Francisco: W.H. Freeman. ISBN 0-7167-3268-8. (İngilizce)
- Santiago Schnell, Philip K. Maini (2004). "A century of enzyme kinetics: Reliability of the KM and vmax estimates". Comments on Theoretical Biology 8: 169–87. DOI:10.1080/08948550302453. http://www.informatics.indiana.edu/schnell/papers/ctb8_169.pdf. (İngilizce)
- Walsh, Christopher (1979). Enzymatic reaction mechanisms. San Francisco: W. H. Freeman. ISBN 0-7167-0070-0. (İngilizce)
- Cleland, William Wallace; Cook, Paul (2007). Enzyme kinetics and mechanism. New York: Garland Science. ISBN 0-8153-4140-7. (İngilizce)
Dış bağlantılar
- Bir enzim ölçümünn animasyonu — Shows effects of manipulating assay conditions.
- MACiE — Enzim mekanizmaları veritabanı.
- ENZYME — Expasy enzim adlandırma sistemi veritabanı.
- ExCatDB — Enzim katalitik mekanizmaları veritabanı.
- BRENDA — Kapsamlı bir enzim veritabanı, substratlar, inhibitörler ve reaksiyon çizimleri bulunmaktadır.
- SABIO-RK — Reaksiyon kinetkileri veritabanı.
- Joseph Kraut Araştırma Grubu, University of California San Diego — Birkaç enzim reaksiyonun mekanizmasının animasyonu.
- Enzim Kinetiğinde sembolizm ve terminoloji — Enzim kinetiğindeki kavramlar ve terminolojinin kapsamlı bir açıklaması.
- Enzim kinetiğine bir giriş — Enzim kinetiği hakkında çevrimiçi, kolay anlaşılır bir eğitsel.
- Enzim kinetiği animasyonlu eğitsel.